Wednesday, April 24, 2024
12:30 PM - 01:30 PM
Goddard Centerwide Town Hall
Goddard Center Director Dr. Makenzie Lystrup will host the next centerwide town hall from the Goddard Institute for Space Studies (GISS) in New York. Topics for discussion will include the latest budget updates, Goddard 2040, the center’s role in the agency’s Artemis program and Moon to Mars strategy, and the importance of GISS and Earth science missions to the Goddard portfolio.
Read more about this event Wednesday, April 24, 2024
02:00 PM - 03:00 PM
SMD Town Hall
Details to follow
Read more about this event Wednesday, April 24, 2024
03:00 PM - 04:30 PM
Scientific Colloquium - BLDG 8
Planet Nine from Outer Space: A Status Update and New Evidence
Konstantin Batygin (California Institute of Technology)
Read more about this event Konstantin Batygin (California Institute of Technology)
Thursday, May 02, 2024
11:10 AM - 11:55 AM
Virtual Chat with the Code 600 Director
Everyone is Welcome!
ACTUAL TIME: 11:10 - 11:55
Read more about this event ACTUAL TIME: 11:10 - 11:55
Wednesday, May 08, 2024
01:00 PM - 04:30 PM
Workshop for ROSES Proposal Writers
SPSO will offer a Workshop for ROSES Proposal Writers. We do this at least annually and each time address new features of the Research Opportunities in Space and Earth Science (ROSES) solicitation. We encourage in-person attendance.
12:45 – 1:00 pm: Sign into virtual meeting, work out kinks, share stories
1:00 – 1:15 pm: What are Research and Analysis (R&A) proposals?
1:15 – 1:45 pm: How are R&A proposals evaluated?
1:45 – 2:00 pm: How do I find R&A opportunities?
2:00 – 2:15 pm: 15-minute break
2:15 – 3:00 pm: What are Goddard’s policies and Best Practices for proposal writers?
3:00 – 3:20 pm: How do I create a budget?
3:20 – 4:35 pm: How do I create a persuasive proposal?
Read more about this event 12:45 – 1:00 pm: Sign into virtual meeting, work out kinks, share stories
1:00 – 1:15 pm: What are Research and Analysis (R&A) proposals?
1:15 – 1:45 pm: How are R&A proposals evaluated?
1:45 – 2:00 pm: How do I find R&A opportunities?
2:00 – 2:15 pm: 15-minute break
2:15 – 3:00 pm: What are Goddard’s policies and Best Practices for proposal writers?
3:00 – 3:20 pm: How do I create a budget?
3:20 – 4:35 pm: How do I create a persuasive proposal?

FEATURED STORY

At 7:26 this morning, BurstCube was successfully deployed from the ISS. The team now awaits the opportunity to communicate with the satellite, some t...
Thursday, April 18, 2024The OSIRIS-Rex Team has been awarded the prestigious award for innovation in aviation, Read more about this
3, 2, 1 … LIFTOFF! A SpaceX Falcon 9 rocket carrying NASA’s PACE (Plankton, Aerosol, Cloud, ocean Ecosystem) spacecraft launched on a SpaceX Falcon 9 ...
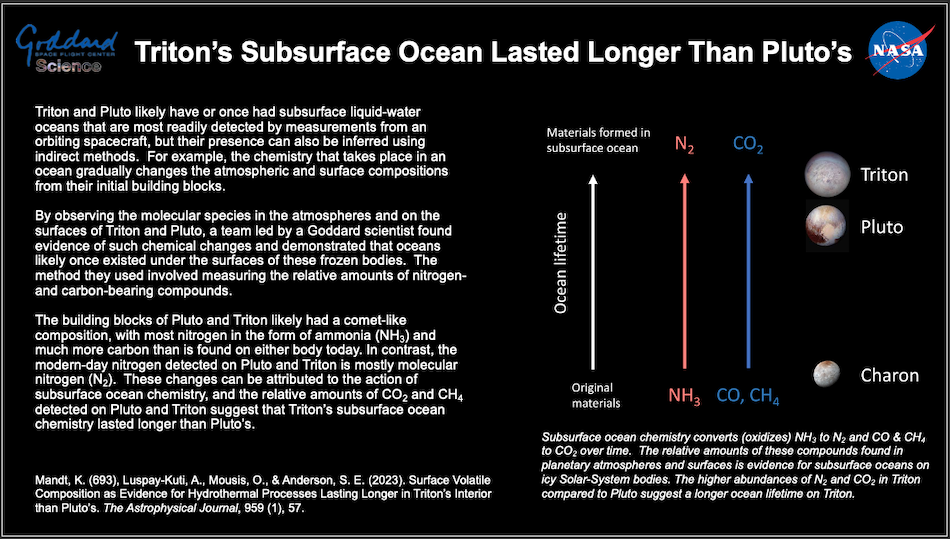
Thursday, February 08, 2024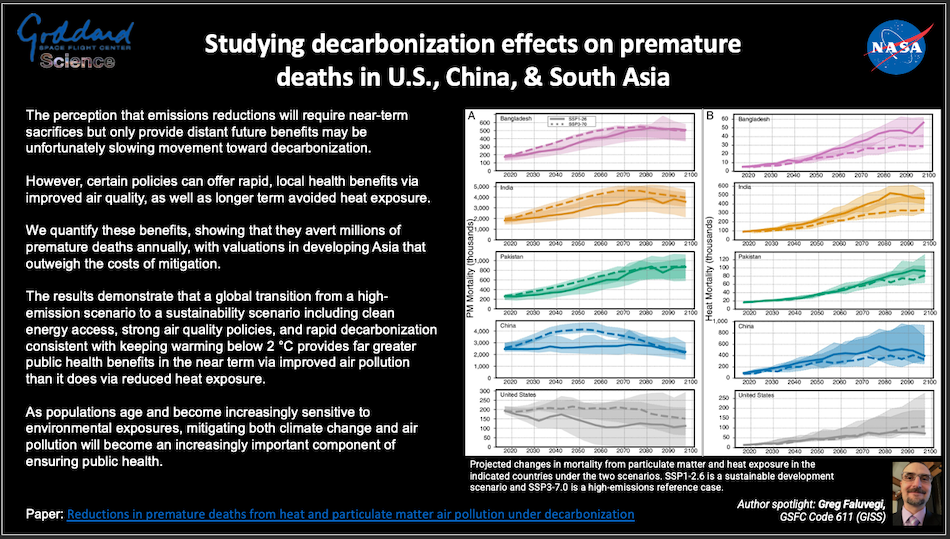


APOD

Earth Observatory Picture

HEASARC
Contact Us
Technical issues with this website may be reported to karen.smale@nasa.gov.
General inquiries about the scientific programs at NASA's Goddard Space Flight Center may be directed to the Center Office of Communications at 1.301.286.8955.
General inquiries about the scientific programs at NASA's Goddard Space Flight Center may be directed to the Center Office of Communications at 1.301.286.8955.